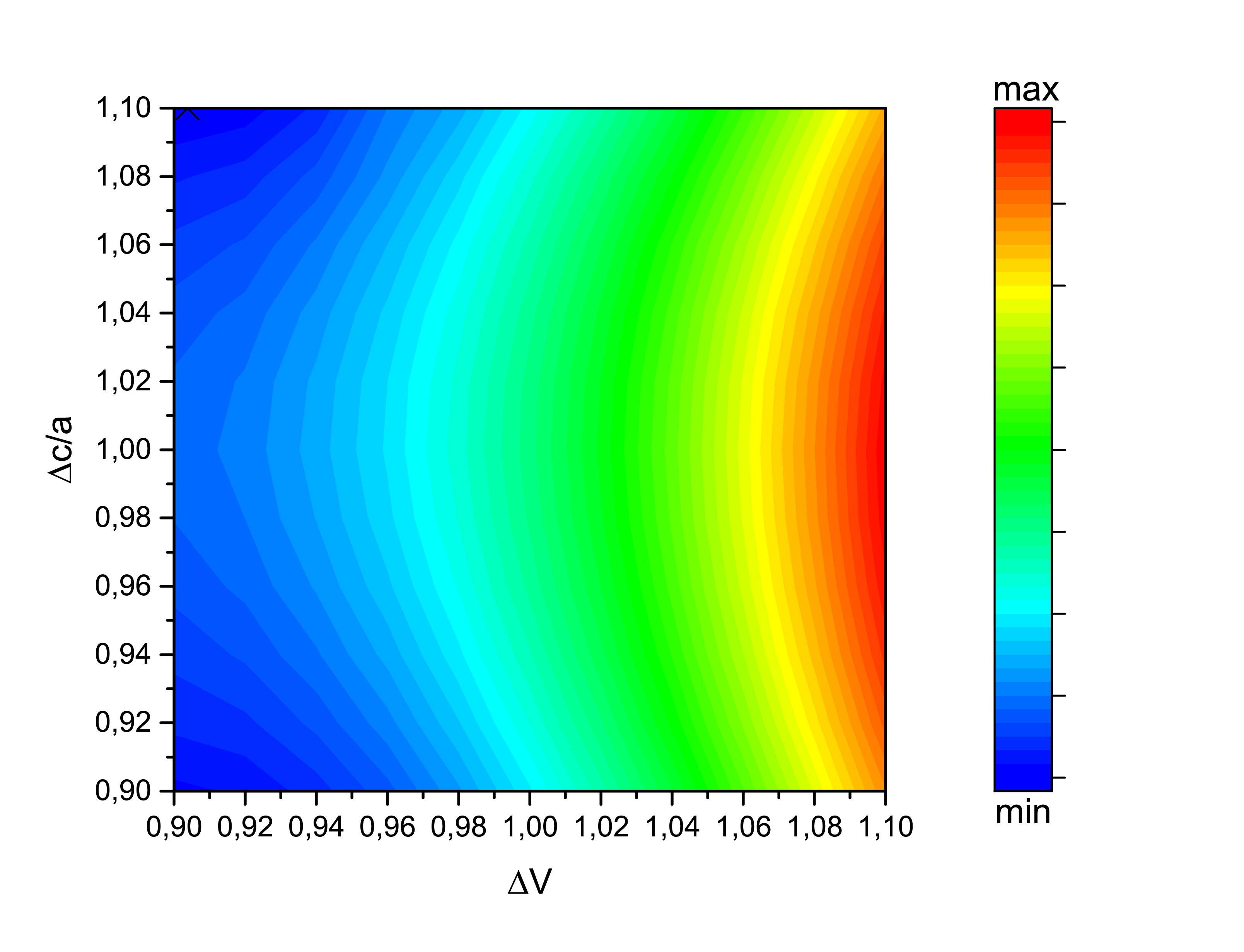
The Vienna Ab initio Simulation Package and the Munich Spin-Polarized Relativistic Korringa-Kohn-Rostoker package use different approaches to calculate the ground state energy of a structure. Even without an ionic relaxation is still important to perform an electronic relaxation before we calculate some of its properties such as its exchange–correlation energies. Beyond that, we can compare the obtained results to the well known experimental properties of the crystalline structure. Variations of 1% from -10% to +10% of the experimental Keshavarz et al. volume and c/a were applied using SPR-KKR. The LDA functional (at the left) could not predict the ground state. Trying the GGA functional (at the right) we reach the conclusion that SPR-KKR may not be the best option to optimize the D0\(_{19}\) Fe\(_{2}\)MnGe structure
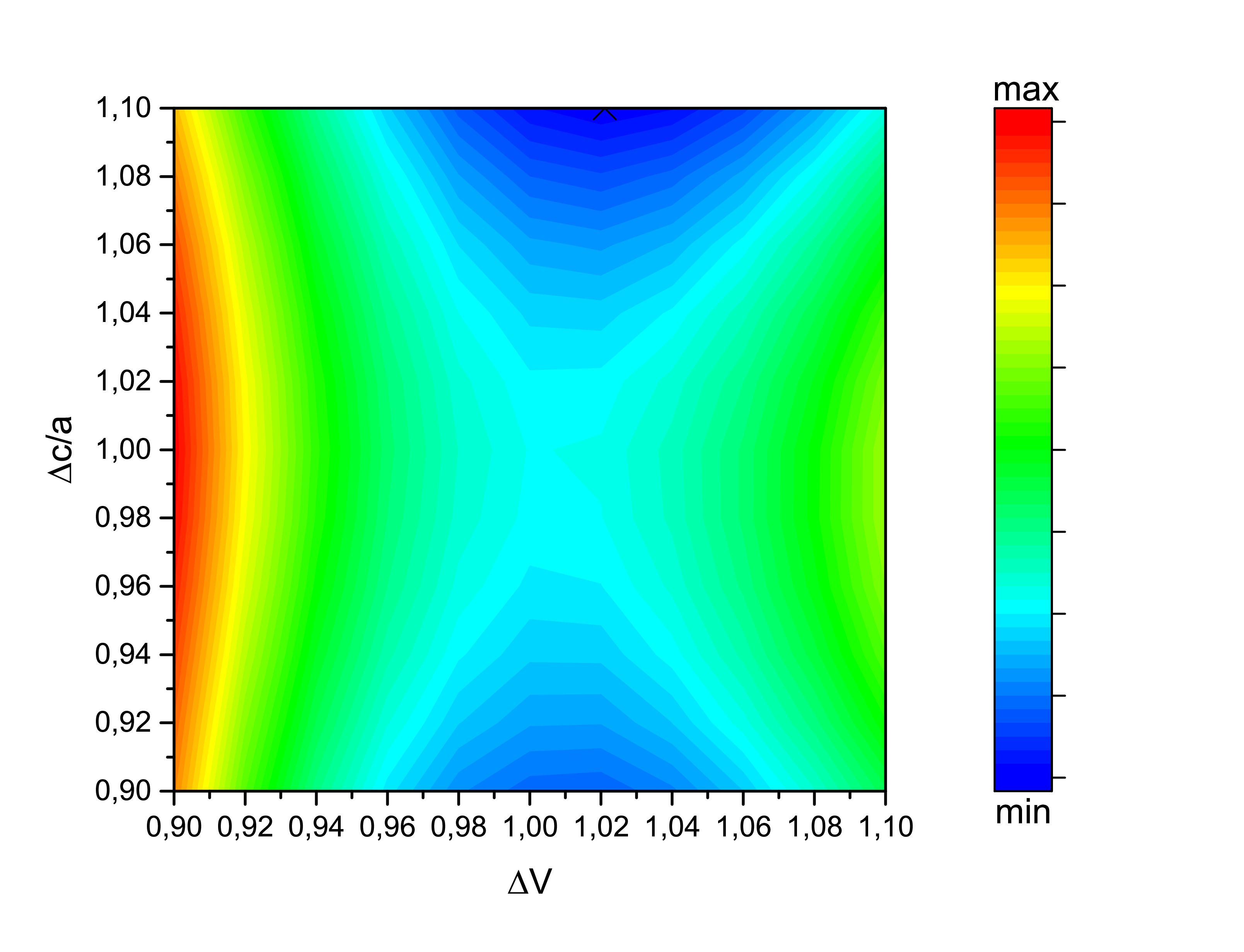
The same structural optimization was calculated using VASP, at left using the LDA functinal and at right using the GGA functional. Despite no energy local or absolute minimum was reached with SPR-KKR, VASP found the ground state energy to be (0.86,1.00) and (0.96,1.00) with LDA and GGA, respectively. While c/a is approximately equal to the experimental one reported by Keshavarz et al., the LDA preddicts the volume to be far away from it. GGA functional is coherent with the experimental values.
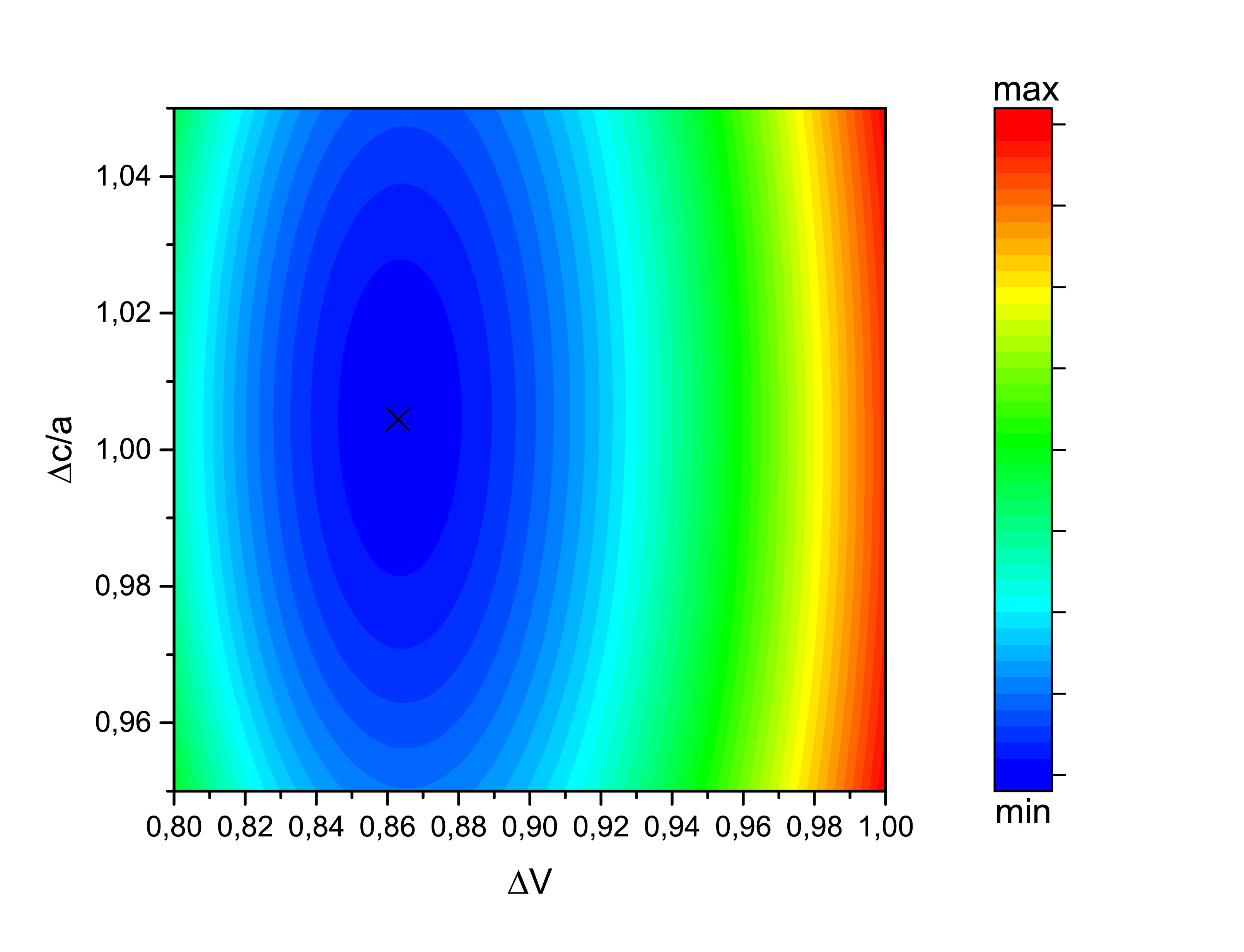

After all, the optimized volumes are V\(_{GGA}\)=96.1 Å\(^{3}\) and V\(_{LDA}\)=86.1 Å\(^{3}\) using VASP. We now can calculate othe properties which may be of our interest.